Malaysia mentakrifkan penyakit jarang jumpa sebagai sebarang penyakit yang mempunyai kekerapan berlaku yang rendah (<1 dalam 4,000 orang), serius dan boleh mengancam nyawa dan/atau menyebabkan ketidakupayaan yang kronik.
Penyakit neurobiromatosis type 1 (NF1) atau Von Recklinghausen turut disenaraikan sebagai penyakit jarang jumpa di dalam Malaysian Orphan Medicines Guideline dalam kategori rare neurological and neuromuscular disease.
NF1 adalah penyakit genetik yang menyebabkan bahagian kulit dan tulang menjadi tidak normal di samping menyebabkan ketumbuhan besar atau kecil di bahagian saraf neural yang boleh berlaku di otak, saraf tunjang dan urat saraf kecil atau besar.
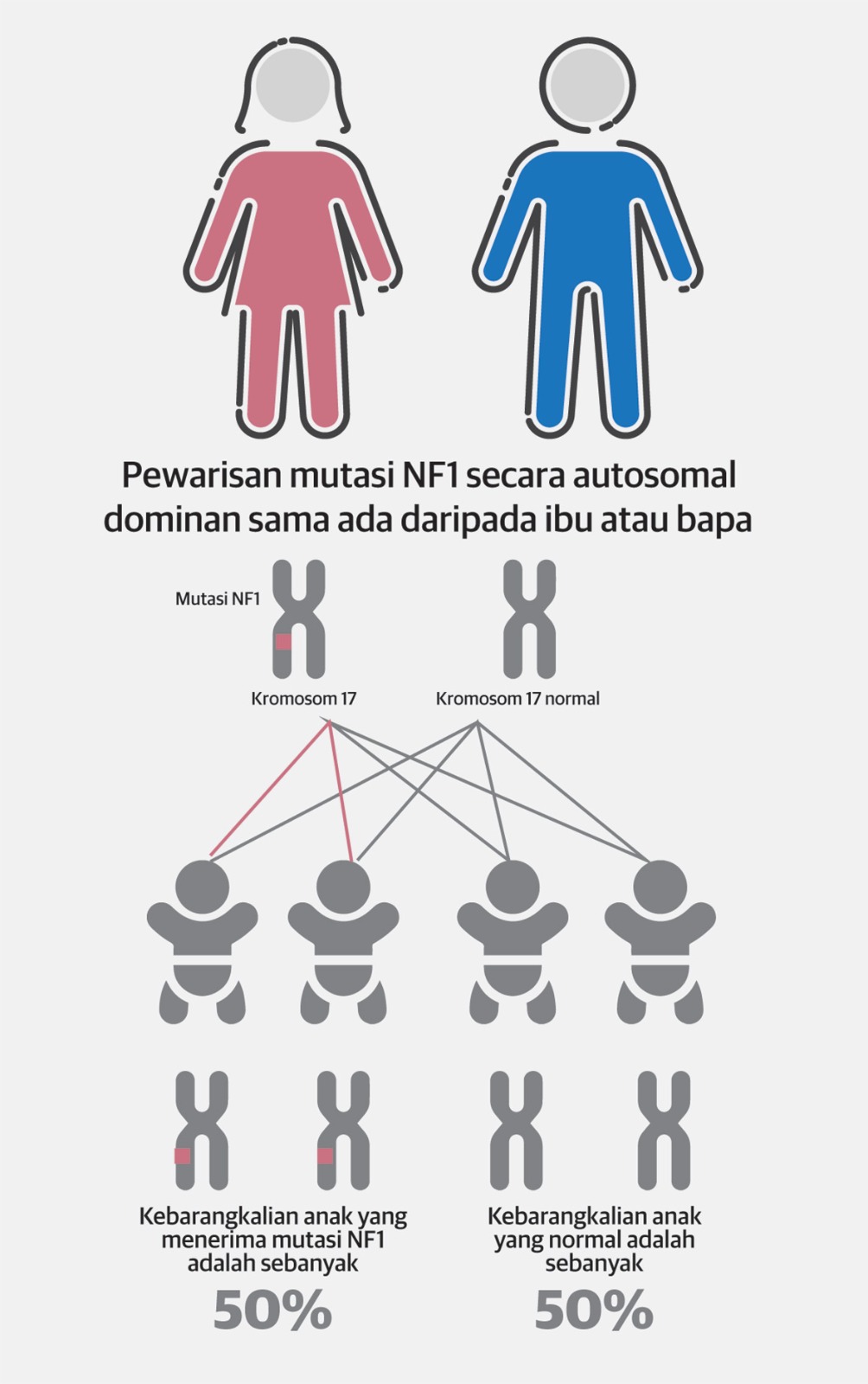
Walaupun NF1 lazimnya diwariskan melalui autosomal dominan kepada pesakit, sekitar 50 peratus daripadanya disebabkan mutasi secara spontan yang berlaku pada keluarga yang tiada sejarah mengalami penyakit tersebut.
Individu yang menghidap NF1 mempunyai 50 peratus peluang untuk mewariskan varian penyebab (patogenik) NF1 kepada keturunan seterusnya, yang mana ciri-ciri NF1 mungkin jauh lebih teruk atau kurang dalam penghidap pediatrik berbanding ibu bapa penghidap NF1.
Apakah punca NF1?
NF1 berlaku disebabkan mutasi pada kromosom nombor 17 yang menghasilkan protein yang dikenali sebagai neurofibromin bagi membantu mempelbagaikan serta mengawal perkembangan saraf.
Apabila mutasi terjadi pada kromosom 17, maka penghasilan protein neurofibromin yang tidak berfungsi seterusnya tidak mampu mengawal pertumbuhan dan pembahagian sel dengan sempurna. Kesannya, ketumbuhan seperti neurofibroma boleh membentuk pada saraf di seluruh badan.
Simptom utama NF1
NF1 boleh dijangka pada individu yang memenuhi dua atau lebih ciri klinikal iaitu kehadiran enam atau lebih tompok berwarna kopi susu (cafe au lait) yang rata di sekitar dada, bahagian belakang dan abdomen, dalam pelbagai saiz; pada individu akil baligh. Kehadiran dua atau lebih neurofibromas atau satu plexiform neurofibromas (PN), dua atau lebih Lisch nodules dan pseudoarthrosis yang ditentusahkan oleh pakar perubatan juga antara simptom utama penyakit NF1.
Jenis Neurofibromatosis
Neurofibromatosis merujuk kepada tiga gangguan genetik berkaitan yang dipanggil NF1, NF2 dan Schwannomatosis. NF1 biasanya didiagnosis pada usia kanak-kanak dan adalah yang paling kerap berlaku (96 peratus) manakala NF2 dan Schwannomatosis selalunya dikesan pada usia remaja. NF2 adalah lebih jarang berlaku berbanding NF1 iaitu sekitar 3 peratus dan Schwannomatois, kurang daripada 1 peratus.
Apa yang berlaku sekiranya pesakit itu menghidap NF1?
Hampir separuh penghidap NF1 mempunyai plexiform neurofibromas (PN) yang menyebabkan kesakitan, gangguan neurologi, ketidakupayaan pembelajaran dan ketidakupayaan kekal. Manisfestasi klinikal yang lebih berpotensi menjadi serius seperti skoliosis (tulang belakang bengkok), tulang rapuh, tekanan darah tinggi dan penyakit pulmonari.
Ketumbuhan boleh muncul di pelbagai kawasan sekitar badan, dalam pelbagai saiz dengan komplikasi tertentu seperti epilepsi dan hilang penglihatan. Kebiasaannya, ia adalah jenis tidak berbahaya (benign) tetapi ada sebahagian tumor ini boleh menjadi lebih agresif menjadi sel kanser.
Adakah ujian genetik penting dalam diagnosis NF1?
Jika pesakit memenuhi kriteria simptom utama NF1, ujian penjujukan genetik adalah penting untuk menentusahkan mutasi yang berlaku. Ia boleh dilihat melalui teknik Next Generation Sequencing, yang mana ia dapat mengenal pasti kehilangan gen NF1 yang terletak di kromosom 17 dan juga varian patogenik yang berupaya menzahirkan simptom-simptom tersebut.
Teknik analisis kromosom juga boleh dilakukan untuk melihat sebarang kehilangan gen itu secara mikro. Jika pesakit menunjukkan hanya satu simptom utama NF1, panel pelbagai gen akan digunakan untuk mengesan beberapa jenis mutasi yang mempunyai korelasi rapat dengan simptom sedia ada pada pesakit.
Ujian genetik adalah penting bagi menentukan jika pewarisan ini secara autosomal dominan, ataupun secara spontan (de novo) dan menentukan jenis varian penyebab yang spesifik.
Apa ibu bapa perlu lakukan jika anak menghidap NF1?
Sesi kaunseling genetik lazimnya diberikan kepada ibu bapa yang menjalani keputusan ujian genetik. Ini adalah penting bagi perancangan keluarga pada masa akan datang. Ini juga untuk memberikan kesedaran bagaimana penyakit ini terjadi dan mengurangkan rasa bersalah ibu bapa yang selalu menyalahkan diri mereka kerana menjadi pembawa mutasi gen ini.
Jika mutasi ini didapati secara autosomal dominan, kebarangkalian setiap anak mendapat mutasi gen NF1 ini adalah sebanyak 50 peratus dan kebarangkalian untuk menjadi normal adalah 50 peratus.
Akan tetapi, jika mutasi ini berlaku secara spontan (kedua ibu bapa adalah pembawa gen normal), ibu bapa tidak perlu risau, kerana kebarangkalian bagi anak seterusnya mendapat mutasi itu adalah rendah.
Kaedah rawatan
Kementerian Kesihatan bersetuju meluluskan penggunaan produk perubatan bagi penyakit jarang jumpa (orphan medicine) Kuselugo dalam bentuk kapsul 10mg dan 25mg yang masing-masing mengandungi bahan aktif selumetinib 10mg dan 25mg untuk indikasi Neurofibromatosis type 1 with inoperable plexiform neurofibromas (PN) pada pesakit paediatrik dengan NF1 berusia 3 tahun ke atas.
Produk Kuselugo telah diberikan status orphan medicine yang diproses secara priority review di mana tempoh penilaian bagi laluan pendaftarannya hanya mengambil 120 hari bekerja sahaja dan merupakan produk pertama yang diluluskan PBKD bagi indikasi NF1.
Kuselogo yang didaftarkan oleh AstraZeneca telah pertama kali diluluskan di Kesatuan Eropah pada Jun 2021 berdasarkan keputusan positif daripada percubaan SPRINT Stratum 1 Fasa II yang mana bahan aktif selumetinib berjaya menyusutkan plexiform neurofibromas dalam 66 peratus pesakit dan menunjukkan peningkatan bermakna secara klinikal dalam gejala berkaitan PN. Melalui akses ubatan berkualiti ini, diharap dapat memberikan sinar harapan buat semua pesakit NF1. – Mingguan Malaysia
Penulis ialah Dr. Noor Akmal Shareela Ismail (Jabatan Biokimia, Fakulti Perubatan UKM) dan Prof. Madya Dr. Nurul Izzaty Hassan (Jabatan Sains Kimia, Fakulti Sains Teknologi, UKM).